
服務熱線:13621695486021-58201756
服務熱線:13621695486021-58201756
1 引 言
喹諾酮類藥物是一類具有抗菌作用的人工合成藥物,具有抗菌譜廣、抗菌力強、毒副作用小、價格低 廉等特點,被廣泛用于治療各種感染性疾病。然而,該類藥物容易導致耐藥菌株的產生,且具有潛在的 致癌性和遺傳毒性,可能對人體健康造成潛在風險。在畜牧和養殖領域,由于不合理用藥、不遵守 休藥期等原因,導致藥物可能會超標殘留于生物體內。為確保動物源性食品質量安全,開發喹諾酮類藥 物殘留的檢測方法至關重要。目前,《中華人民共和國藥典》( 2015 年版) 中針對喹諾酮類藥物的檢測 方法主要為分光光度法和高效液相色譜法,報道的方法包括高效液相色譜-串聯質譜法、毛 細管電泳法、酶聯免疫法和膠體金法等。其中,分光光度法、酶聯免疫法和膠體金法操作簡 單,但選擇性較差,靈敏度較低; 毛細管電泳法快速高效,但分離重現性較差。高效液相色譜-串聯質譜 法兼具靈敏度高、特異性好等優勢,是目前喹諾酮類藥物殘留檢測的常用方法。
2 實驗部分
2.1 儀器與試劑
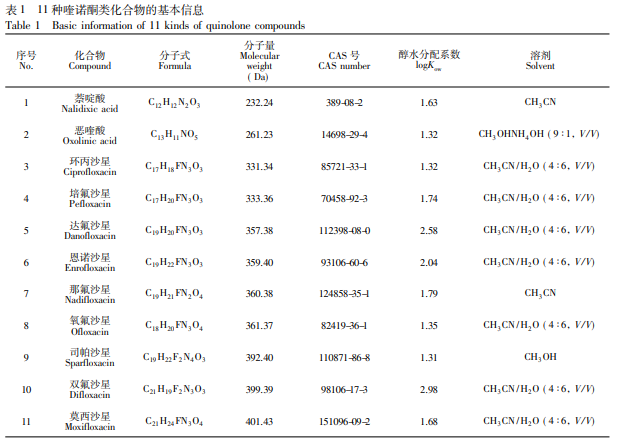
ACQUITY 超高效液相色譜儀( 美國 Waters 公司) ; QTRAP 6500+三重四極桿/復合線性離子阱質譜 儀、Analyst 和 MultiQuant 數據處理系統( 美國 SCIEX 公司) ; Milli-Q Integral 5 型超純水儀( 美國 Merck Millipore 公司) ; AB204-S 型電子天平( 瑞士 Mettler Toledo 公司) ; CR 21N 型高速冷凍離心機( 日本 Hitachi 公司) ; MS3 型渦旋振蕩器( 德國 IKA 公司) ; 昆山舒美KQ-600型超聲波清洗器( 昆山市超聲儀器有限 公司) 。 甲醇、乙腈和四氫呋喃( 色譜純,美國 Fisher 公司) ; 正戊醇、正己醇、正辛醇、正癸醇、正十一醇和正十 二醇( 百靈威科技有限公司) ; 正庚醇和正壬醇( 日本 TCI 公司) 。11 種喹諾酮類化合物標準物質: 萘啶酸 ( 99.5%) 、惡喹酸( 98.0%) 、鹽酸環丙沙星( 95.0%) 、達氟沙星( 97.5%) 、恩諾沙星( 98.0%) 、那氟沙星( 99. 0%) 、司帕沙星( 99.0%) 、鹽酸雙氟沙星( 97.5%) 和鹽酸莫西沙星( 96.1%) 購自德國 Dr. Ehrenstorfer 公司) ; 培氟沙星( 98.0%,加拿大 TRC 公司) ; 氧氟沙星( 98.8%,中國食品藥品檢定研究院) 。11 種喹諾酮類化 合物的基本信息見表 1。
2.2 實驗方法
2.2.1 標準溶液的配制
準確稱取 11 種喹諾酮類化合物標準物質各 10 mg( 精確至 0.1 mg) ,分別用表 1 中對應的溶劑溶解并定容至 10 mL,配制成濃度為 1 mg /mL 的標準儲備溶液。分別取 11 種喹諾酮類化合 物標準儲備溶液,配制成濃度為 20 μg /mL 的混合標準儲備溶液,4℃保存。使用時以甲醇逐級稀釋成系列 濃度的標準工作溶液,現用現配。
2.2.2 超分子溶劑的制備
移取 2.5 mL 正己醇和 8 mL 四氫呋喃,迅速注入 50 mL 離心管中,加入 29.5 mL 水,以 420 r/min 磁力攪拌 3 min,3000 r/min 離心 10 min,用注射器移取上層有機相于適宜容器 中,4℃密封保存。
2.2.3 樣品處理
采用活體取血法取血后立即移取 0.1 mL 血液樣品,置于預先加入 0.2 mL 超分子溶劑的 2 mL 聚丙烯離心管中,渦旋振蕩 3 min,以 5000 r/min 離心 10 min。移取上層液體 50 μL 于0.3 mL微量進 樣瓶中,加入 50 μL 甲醇混勻,過 0.22 μm 微孔濾膜后,待超高效液相色譜-三重四極桿/復合線性離子阱質 譜測定。
2.2.4 分析條件
( 1) 色譜柱 ACQUITY UPLC BEH C18( 50 mm×2.1 mm,1.7 μm) ; 流速: 0.35 mL/min; 流動相: 0.15%甲酸水溶液( A) 和乙腈( B) 。
( 2) 梯度洗脫程序 0~1.8 min,10% ~30% B; 1.8~2.2 min, 30%~90% B; 2.2~3.2 min,90% B; 3.2~3.5 min,90% ~10% B; 3.5~5.0 min,10% B; 柱溫: 30℃; 進樣 量: 2 μL。
( 3) 電噴霧質譜 電噴霧離子源; 正離子掃描; 離子源溫度: 150℃; 噴霧電壓: 5500 V; 氣簾 氣流速: 30 psi,霧化氣流速: 60 psi,脫溶劑氣流速: 60 psi; 脫溶劑氣溫度: 600℃; 碰撞氣設置: 高。
( 4) 多反應監測-信息關聯采集-增強子離子掃描復合模式( MRM-IDA-EPI) 掃描范圍: m/z 50~415; 掃描速 度: 20000 Da /s; 碰撞能量: ( 35±15) V。
3 結果與討論
( 1) 烷基醇種類 選用兩親性有機溶劑四氫呋喃分別與碳原子數 C5 ~ C12的烷基醇 ( 正戊醇、正己醇、正庚醇、正辛醇、正壬醇、正癸醇、正十一醇和正十二醇) 和水組合形成超分子溶劑,對 11 種喹諾酮類化合物進行萃取。11 種物質的萃取率隨著烷基醇碳原子數的增加,整體呈現下降的趨勢。反向膠束超分子溶劑萃取作用力主要包括烷基鏈的疏水相互作用和親水頭基的氫鍵作用。 當烷基醇碳鏈較長時,疏水相互作用占優勢,適合萃取極性小的物質; 當烷基醇碳鏈較短時,氫鍵作用占優 勢,適合萃取極性大的物質。隨著碳鏈長度的減小,超分子溶劑相給質子能力會隨之提高,從而使分子 間作用力增強,喹諾酮類化合物萃取率增加。總體而言,正己醇具有較好的萃取效果( 萃取率為 65.0% ~ 113.1%) ,而正辛醇及碳鏈更長的烷基醇的萃取率較低。另一方面,喹諾酮類物質的化學結構中含有苯環、 哌嗪環、環丙烷等結構,導致其空間位阻變大,短鏈烷基醇更容易克服空間位阻而與氫鍵位點結合,有助于 提高萃取效率。因此,最終選擇正己醇用于后續優化試驗。
( 2) 烷基醇用量 保持超分子溶劑總體積為 40 mL,四氫呋喃用量為4 mL,分別選取正己醇用量為 1、1.5、2、2.5、3、3.5 和 4 mL 進行了考察。當正己醇用量大于 2.5 mL 時,樣品可獲得較好的萃取效率。
( 3) 四氫呋喃用量 考察了四氫呋喃 用量為 2、4、6、8、10 和 12 mL( 保持超分子溶劑總體積為 40 mL 及正己醇用量為 2.5 mL) 的萃取情況 ,結果表明,四氫呋喃用量在 8~10 mL 時可獲得較好的萃取效果。
( 4) 渦旋時間 在超分子溶劑 萃取過程中,通過渦旋振蕩可以加速超分子溶劑和目標化合物之間充分混合,促進傳質過程。比較了渦旋 時間為 1、2、3、5、7 和 9 min 時的萃取效果 ,當渦旋時間在 3~7 min 時,可獲得較理想的萃取效果。 通過單因素試驗確定各參數的大致優化范圍后,通過正交試驗對超分子溶劑萃取條件進行進一步優化。
4 結 論
建立了超分子溶劑分散液液微萃取結合超高效液相色譜-三重四極桿/復合線性離子阱質譜測定血液 中 11 種喹諾酮類藥物殘留的分析方法,通過單因素和正交試驗考察了影響萃取效果的烷基醇種類與用 量、四氫呋喃用量等關鍵因素,并進行了相關的統計學分析。在最佳條件下,利用多反應監測-信息關聯采 集-增強子離子掃描復合模式測定 11 種待測化合物的檢出限為 0.1~0.8 μg /L,定量限為 0.4~2.0 μg /L; 在 低中高 3 個加標水平下的平均回收率為 70.8% ~115.2%,相對標準偏差為 3.0% ~11.5%。
免責聲明:文章僅供學習和交流,如涉及作品版權問題需要我方刪除,請聯系我們,我們會在第一時間進行處理。
產品中心 | 技術支持 | 下載中心 | 在線留言 | 合作伙伴 | 新聞動態 | 關于舒美 | 聯系我們 |

Copyright © 2002-2020 上海蟻霖科學儀器有限公司 版權所有
備案號: 滬ICP備19002068號
服務熱線:13621695486
售后電話:021-58201756
公司郵箱:[email protected]
公司地址:上海浦東新區金高路2131弄17號401室
 掃一掃進入手機官網
掃一掃進入手機官網
 掃一掃關注我們
掃一掃關注我們
